<<ウェブ公開版操作マニュアル>>
プロテオーム解析のための二次元電気泳動標準操作法およびインゲル消化操作法
(東京都老人総合研究所産学公連携プロテオーム共同研究センター標準操作法)
(2009年2月12日 改訂版)
ここに記載された方法に従って実施された研究の成果を論文等で発表される際は、和文の論文の場合には
「固定化pH勾配ゲルストリップを用いた二次元電気泳動」
戸田年総, 生物物理化学, 41巻, 4号, 169-172 (1997)を引用して下さい。英文の論文の場合には、
"Standardization of protocol for
Immobiline 2-D PAGE and construction of 2-D PAGE protein database on
World Wide Web home page" Jpn. J.
Electrophoresis, 41(1), 13-20 (1997)
を引用して下さい。
なおこれらの論文には、方法の細部にわたっての記載はされてはおりませんので、和文の場合にはこの『ウェブ公開操作マニュアル』のURLを、英文の場合には
英語版公開マニュアルのURL (http://www.proteome.jp/2D/2DE_method.html)
を論文上で引用していただきますようお願い致します。
また、「13.染色されたゲルからPVDF膜へのタンパク質転写」については、"Proteomic analysis of Epstein-Barr
virus-transformed human B-lymphoblastoid cell lines before and after
immortalization" Electrophoresis, 21(9), 1814-1822 (2000)
で報告しておりますので、こちらを引用して下さい。
東京都健康長寿医療センター研究所
(東京都老人総合研究所)
産学公連携プロテオーム共同研究センター
センター長
戸田 年総
1.二次元電気泳動に必要な装置
1-1.固定化pH勾配等電点電気泳動を行うための装置は、 バイオラッド、 アナテック、 GEヘルスケアバイオサイエンス、などから市販されています。ここではアナテックの装置を例にとって取り扱い
方
を説明しますが、原理的な 部分は各社共通ですので、他の装置を用いた場合でも、基本的には同様の条件で行うことができます。
1-2.等電点電気泳動では、徐々に電圧を上昇させながら泳動を行うために、プログラム設定ができる直流高圧電源が必要です。
(これも上記の各社が取り扱っています。)
1-3.二次元目のSDS電気泳動装置は、スラブゲルの上端に一次元目のゲルストリップが横方向に入る寸法のものであればどのようなものでも構いません
が、できるだけ
スマイリングが起こらないようにするために、ゲル板全体が電極液に浸って温度を均一に保てるものが良いでしょう。これも、上記各社から発売されています
(バイオ
ラッド PROTEAN II、 アナテック クールホレスター IPG-IEF、 GEヘルスケア Hoefer SE 600 Ruby)。
1-4.蛍光画像の読み取り装置としては、発光ダイオードを光源とし
CCDカメラで撮影するものと、レ−ザー光源で励起しスキャナーで取り込む方式の物があります。(当センターでは Bio-Rad の Pharos
FX を使用しています。)
1-4.画像解析を行うためのソフトウエアとしては、GE の Image Master 2D や バイオラッド の PDQuest,
島津製作所が扱っている Progenesis などがあります。(当センターでは PDQuest を使用しています。)
2.固定化pH勾配等電点電気泳動用ストリップゲル(ドライストリップゲル)
固定化pH勾配等電点電気泳動用ストリップゲルはGE
ヘルスケアとバイオラッド
から、様々な長さや pH 領域のものが乾燥した状態で販売されています。
3.試薬類
以下のものを使用します。ただしこれらは必ずしも全てが必要ではなく、泳動の条件や染色法の選択によっては不要なものもあります。(参考までに、当センターで使用しているものを括弧内に示しますが、これより純度が高いものであれば、他のメーカーのものでもかまいません)
トリス(TRIZA BASE, Sigma T-1503)、トリシン(Sigma T-5816)、グリシン(ナカライテスク
17141-95)、グリセロール(和光純薬 075-00616)、オレンジG(Sigma O3756)、BPB(Aldolich
11,439-1)、尿素(ナカライテスク 35940-81)、チオ尿素(Sigma T-7875)、SDS(ナカライテスク
31607-65)、Triton X-100(Sigma C-5070) 、CHAPS、SB3-10(Sulfobetaine 10、Amresco
J548-10G)(代理店:コスモ・バイオ)、βメルカプトエタノール、DTT(Sigma
D-0632)、メタノール(和光試薬特級)、酢酸(和光試薬特級)、CBB染色試薬(和光 Quick-CBB)、SYPRO
Ruby(invitrogen
S12000)、Pro-Q Diamond (invitrogen
P33300)、低粘度のシリコンオイル(
信越シリコーン KF-96L-5cs)、その他。
4.タンパク質抽出液の調製
4-1.培養細胞の場合:
浮遊した状態で培養されたものであれば、あらかじめ風袋重量を測定しておいた遠心管に移し、5,000 rpm
で5分間程度遠心して細胞を集め、氷冷したPBSで再懸濁して遠心する形で3回洗浄して、培地に由来するタンパク質を除く。最後にできるだけ完全にPBS
を除いた後で重量を測定し、細胞重量を求める。
接着して増える細胞の場合、培地を除いて氷冷したPBSで2回容器内壁を洗浄し、氷上でプラスチックスクレーパーを用いて細胞を掻き集める。これを、あ
らかじめ風袋重量を測定しておいた遠心管に移し、5,000 rpm
で5分間程度遠心して細胞を集め、できるだけ完全にPBSを除いた後で重量を測定し、細胞重量を求める。
この細胞に、重量の4倍量の抽出バッファー(A,BもしくはC*)を加えて、氷冷下で超音波破砕する。
15,000 rpm で5分間遠心し、上清を少量ずつ分注して、-80℃に保存する。
4-2.実験動物などの組織の場合:
メスの刃先や眼科用のハサミなどで細かく砕いた後にあらかじめ風袋重量を測定しておいた遠心管に移し、組織重量を測定する
組織重量の4倍量の抽出バッファー(A,BもしくはC*)を加え、眼科用のハサミで組織を細かく砕いた後に、氷冷下で超音波破砕
する(5秒間 x10回〜20回程度)。
15,000 rpm
で5分間遠心し、上清を少量ずつ分注して、-80℃に保存する。(膜タンパクの一部には、凍結することによって不溶化を起し、二次元電気泳動のパターンか
らスポットが消失することがあるので、膜タンパクを取り扱う時には、凍結を避けることが望ましい。
一般に、凍結保存されたサンプルは使用直前に融解し、40℃で5分間程度インキュベートした後に泳動を行う。
抽出バッファーA
For cytosolic/hydrophilic proteins
Urea 0.51 g
10%(w/v) SDS 0.02 ml
20%(v/v) Triton X-100 0.10 ml
DTT 0.01 g
Pharmalyte 3-10 0.02 ml
Milli-Q water up to 1.00 ml
抽出バッファーB
For membrane/particulate-bound proteins
Urea 0.30 g
thiourea 0.15 g
20%(w/v) CHAPS 0.10 ml
20%(w/v) SB3-10 0.10 ml
DTT 0.01 g
Pharmalyte 3-10 0.02 ml
Milli-Q water up to 1.00 ml
<<Option>>これらのバッファーの条件下では、ほとんどのプロテアーゼやホス
ファターゼは失活ると思われますが、分解を完全に抑えるためには、さらに下記の阻害剤を加えることが望ましい。
Sigma Protease Inhibitor Cocktail 10 ul
Sigma Phospahatase Inhibitor I 5 ul
Sigma Phospahatase Inhibitor II 5 ul
抽出バッファーC(Cy3/5-labeling、IC3/5-labeling 用)
For IC3/5-OSu labeling
Urea 0.51 g
10%(w/v) SDS 0.02 ml
20%(v/v) Triton X-100 0.10 ml
0.2 M HEPES, pH 8.0 0.05 ml
Mill-Q water up to 1.00 ml
5.タンパク質の脱塩濃縮(Option):
タンパク質の脱塩濃縮は、CBB染色を行う場合や微量発現タンパク質を検出する場合、質量分析等を高感度で行う必要がある場合などに、必要に応じて行
う。
TCA沈殿法、アセトン沈殿法、凍結乾燥法などさまざまな方法があるが、最もロスが少なく、一般的な方法は限外濾過膜法である。ここでは、ミリポアの
ULTRAFREE UFV5BGC25 を用いた方法を示す。
5-1.ULTRAFREE UFV5BGC25 の上部サンプル容器に、Milli-Q 水 0.5 ml を入れ、卓上冷却遠心機でほぼ
10,000 xg になる回転数にセットし、10 分間遠心する。
5-2.上部、下部両方の液を除き、上部サンプル容器にサンプルを入れる(最大で 0.5 ml)。
5-3.卓上冷却遠心機で10,000 xg 、30 分間遠心する。
5-4.下部サンプル容器の濾液を別のチューブに移し、上部サンプル容器に2次元電気泳動様のサンプル調製用(タンパク質抽出用)バッファーを加えれ全
量を約 0.5 ml に戻す。(単に濃縮だけで、脱塩を行う必要がなければ、バッファーを加える必要はない。その時は5−7に進む。)
5-5.前回の遠心の時の濃縮の具合を見て、遠心時間を調節し、10,000 xg で再度遠心する。
5-6.最初のサンプルの塩濃度が高い場合には、この操作を数回繰り返す。
5-7.適度の量に濃縮されたら、上部サンプル容器の濃縮液を、別のチューブに移して、次の固定化pH勾配等電点電気泳動に使用する。
6.固定化pH勾配等電点電気泳動
6-1.まず最初に、固定化pH勾配等電点電気泳動用の乾燥ストリップゲルを膨潤するためのバッファーを調製する。変性剤や界面活性剤の種類や濃度は、
分離するサンプルの種類によって多少変える必要があるが、老人研では、通常、下記の組成の膨潤液を使用しています。
乾燥ストリップゲル8本を膨潤する時の膨潤液調製法
urea 14.4 g
thiourea 6.1 g
dithiothreitol 0.08 g
Pharmalyte 3-10 0.4 ml
0.1 M acetic acid 1.0 ml
0.1%(w/v) Orange G 1.0 ml
20%(v/v) Triton X-100 4.0 ml
純水を加えて全量を 40 ml とする。
6-2.ゲル1本当たり 5 ml
の膨潤液を容器(アクリル管にシリコンゴム栓をしたもの、もしくは専用の膨潤トレー)に入れ、乾燥ストリップゲルの保護フィルムを剥がして、膨潤液に浸し
ます(通常はゲル面を上向きにする)。
6-3.室温もしくは20℃のチャンバーの中で8時間〜1夜インキュベートし、ゲルを完全に膨潤する。
6-4.膨潤されたストリップゲルを、下図のように濾紙の上に立てて、余分の液を取り除く(約1分間)。
6-5.固定化pH勾配等電点電気泳動装置の温度を20℃に設定し、冷却台の上に泳動用のトレーを置く。
6-6.少量の低粘度シリコンオイル(信越化学の KF-96L-5CS
が最適)をトレーに入れ、泡が入らないように注意しながら、ストリップ固定板を密着させる。
6-7.さらに、ストリップ固定板の上に、少量の低粘度シリコンオイルを流し、それぞれの溝に合わせて、膨潤されたストリップゲルを並べます(アナテッ
クの装置ではゲル面を上にします)。
6-8.陽極側の電極濾紙は純水(Milli-Q 水)に、陰極側の電極濾紙は 5 mg/ml の DTT
を含む純水に浸し、余分の液を濾紙で吸い取って、ゲルの両端の白金電極を接触させる位置に置き、ピンセットで軽く押えてゲルに密着させる。
6-9.サンプル濾紙を、サンプル量に合わせて切り(アナテックの標準品は約18〜20μlのサンプル用)、パラフィルムの上に置き、サンプル(タンパ
ク質抽出液)をしみ込ませて、ピンセットでゲルの端の電極濾紙の近くに置く。なお、通常サンプル濾紙は陰極側に置く方がベターであるが、pH6-11、
pH6-9
(GEヘルスケアライフサイエンス)や pH7-10
(バイオ・ラッドラボラトリーズ)など塩基性領域のストリップの場合には、陽極側に置いた方がテーリングが少なくなる。
6-10.白金電極を、電極濾紙に接触する位置に装着し、低粘度シリコンオイルを注いで、ゲルおよび電極濾紙全体をオイルでカバーし、フタを閉める。
6-11.電源を接続し、pH4-7などの酸性pH領域、あるいは
pH3-10などの広領域のストリップゲルの場合、通常、下記のプログラムで等電点電気泳動を行なう。
(ゲルの長さが17〜18cmのとき)
Step Voltage Time
1 500 V 2 h
2 700 V 1 h
3 1000 V 1 h
4 1500 V 1 h
5 2000 V 1 h
6 2500 V 1 h
7 3000 V 1 h
8 3500 V 10 h
pH6-11、pH6-9 (GEヘルスケアバイオサイエンス)や pH7-10
(バイオラッド)など、塩基性領域のストリップを使用し、陽極側にサンプルをアプライした場合には、下記のプログラムを用いる。
(ゲルの長さが17〜18cmのとき)
Step Voltage Time
1 350 V 8 h
2 500 V 1 h
3 1000 V 1 h
4 1500 V 1 h
5 2000 V 1 h
6 2500 V 1 h
7 3000 V 1 h
8 3500 V 3 h
なお、これらの設定は、サンプルの状態やアプライ量、ゲルの種類、使用する装置によって最適化する必要があります。
ゲルの長さが異なる場合や、電圧勾配モードの電源を使用する場合、低電力モードで泳動する場合など、詳細な条件設定については、別のページ(リンク)
で順次ご紹介する予定ですが、
最終的には各自で最適化をお願い致します。
7.SDS処理と還元アルキル化
7-1.1次元目の等電点電気泳動が終わる直前に、下記の組成のSDS処理液を調製する。(分量は8本の場合)
urea 14.5 g
dithiothreitol 0.2 g
0.5 M Tris-HCl, pH 6.8 2.0 ml
10%(w/v) SDS stock 8.0 ml
0.1%(w/v) BPB 1.0 ml
60%(v/v) glycerol 20.0 ml
7-2.ストリップゲルを取り出し、純水中で軽くすすいでオイルを除いた後に、SDS処理液中に浸し、30分間振盪しながらインキュベートする。
7-3.還元アルキル化は、単に画像解析を行うだけなら必要ありませんが、質量分析を行う予定がある場合には必須です。
下記の試薬を調製し、この中で 20分間インキュベート後、直ちに2次元目のSDS電気泳動を開始する。
0.5 M Tris-HCl, pH 6.8 2.0 ml
10%(w/v) SDS stock 8.0 ml
0.1%(w/v) BPB 1.0 ml
60%(v/v) glycerol 20.0 ml
Milli-Q water 9.0 ml
iodoacetamide 1.8 g
(反応) Protein-SH + ICH2CONH2 --> Protein-S-CH2-CO-NH2
(直ちに2次元目のSDS電気泳動を行うことができない時には、乾燥しないようにサランラップに包んで
-80℃の超低温槽内で保存する)
8.SDS電気泳動
8-1.ゲルの調製(7.5%T, 3%C, トリス/トリシンバッファー系用)
Anatech社製のゲル作成容器(ゲルメーカー37/200)を用いてゲル板4枚を同時に作成する場合、下記の(1)
の試薬を混合溶解し減圧脱気し、氷上で5分間ほど冷却した後で(2) の試薬を混合し、直ちにゲル作成容器に注ぐ。
ゲルの上端をフラットにするために、ゲルモノマー液の上部に少量の精製水を重層し、一晩静置する。(アナテック製のフラットコームを使用する場合には、
水を重層する必要はない)
(1)
35 %(w/v) acrylamide 55 ml
2 %(w/v) BIS-acrylamide 30 ml
1.5 M Tris-HCl, pH 8.8 65 ml
60%(v/v) glycerol 47 ml
pure water up to 260 ml
(2)
10 %(w/v) SDS 2.6 ml
10 %(w/v) ammonium persulfate 1.3 ml
TEMED 0.13 ml
このゲルを用い、トリス/トリシンバッファー系の電極液で泳動すると、おおよそ分子量1万から20万までのタンパク質を分析することができる。
(補足:予めガラス板をシリコナイスしておくと、泳動後にゲルを取り出す際にガラス板からはがれ易くなり、ゲルを破損することが少なくなる。シリコ
ナイズは以下の操作で行う。(1)キムワイプにエタノールをしみ込ませてカラス表面を拭く、(2)ガラス表面にSigmacote
(Sigma-Aldrich
SL-2)を滴下しキムワイプで全体に広げる、(3)乾燥後、キムワイプにエタノールをしみ込ませてもう一度ガラス表面を拭き上げる。なお、この操作は必ず
ドラフトの中で、手袋を着用して行なって下さい。一度シリコナイズを行なうと数回〜十数回は効果が続きます)
(アナテック・クイックランなどのMini-slab電気泳動装置用に 8%T, pH 8.6 のゲルを作成する場合にはこちらをご覧ください。)
8-2.ゲル板の上端の液を除いて、その上に下図のようにストリップゲルを置き、サメの歯状の櫛で押えて、ゲル板とストリップゲルの間を密着させる。
(このときの押さえ加減が重要です)
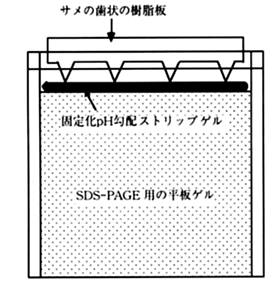
8-3.あらかじめ、陽極液を入れておいた泳動槽にゲル板をセットし、陰極液を満たして、泳動を開始する。
(電極液の調製法および通電条件は下記の通りです)
10x 濃度の陽極液の調製法
Tris 242.2 g を約700 ml の純水に溶かし、6規定の塩酸で pH 8.8 に合わせ、全量を 1,000 ml
とする。
5x 濃度の陰極液の調製法
Tris 60.5 g、Tricine 89.5 g、SDS 5.0 g を純水に溶かし、全量を 1,000 ml にする。(pH
は合わせない)
通電条件:ゲル板1枚当たり 30 mA
程度で、約3時間泳動すると、BPBのバンドがほぼ先端付近に来るので、ちょうど先端に来た時に通電を止める。
9.ゲルの染色
ゲルの高感度染色法には、リン酸化蛋白質を特異的に検出することができるPro-Q
Diamond蛍光染色法と、全ての蛋白質を非特異的に検出するためのSYPRO Ryby
染色法などがあるが、1枚のゲルをこれらの色素で逐次染色して、両者のパターンを比較することもできる。ここでは、逐次染色法について述べる。
SYPRO Ruby 染色だけを行なう時は、9−1から9−7に進んで下さい。
【Pro-Q Diamond 染色】
9-1. 染色用の容器に、50%メタノール/10%酢酸を 200ml
入れ、この中に、ガラス板から取り出したゲルを入れて30分間以上振盪する。
(SYPRO Ruby 染色のみを行なう時は、ここから9−7に進んで下さい)
9-2. 固定液を新しいものに交換し、さらに30分間以上振盪する。(この固定液中で1晩ゲルを保存することもできます)
9-3. 固定液を捨て、Milli-Q 水に交換して10分間震盪する。(この水洗の行程も2回繰り返す)
9-4. 水を捨て、Pro-Q Diamond 染色試薬(ラージゲルの場合 100 ml、ミニゲルでは 50
ml以上)を加えて、暗所(アルミホイルで遮光)で1.5
時間〜2時間、穏やかにゆすりながらインキュベートする。(Onernightでの染色はバックを高くするので、できれば避ける)
9-5. Pro-Q Diamond染色試薬は数回再利用できますので元の容器に戻し、下記の脱色液中で30分間 x
3回以上(バックグラウンドが高い場合は、オーバーナイト)振盪する。(Pro-Q
Diamond染色試薬は冷蔵庫に保存する)
脱色液の調製: Milli-Q 水 750 ml に1M 酢酸ナトリウム(pH 4.0 ) 50 ml 、アセトニトリル 200 ml
を加えて混和する。
9-6. Molecular Imager FX (Bio-Rad
)などのレーザー励起型-蛍光スキャナーや、FluoroPhoreStar3000
(アナテック)などのダイオード励起型-冷却CCD蛍光撮像-スポットポインティング装置でPro-Q
Diamond蛍光染色画像を取り込む(Pro-Q Diamond
は、ガラス板を汚染するので、使用後のサンプルトレーなどは、洗剤でよく洗うようにして下さい)。
SYPRO Ruby 染色を行なわずにこのままゲルを保存する場合には、7%酢酸中で保存する。
【SYPRO Ruby 染色】
9-7. Pro-Q Diamond で染色をせずに直接SYPRO Ruby で染色する場合は、ゲルを
50%メタノール/10%酢酸(ラージゲルの場合 200 ml、ミニゲルで は100
ml 以上)中で30分間以上振盪後、10%メタノール/7%酢酸中で、さらに30分間振盪する。 あらかじめ Pro-Q Diamond
で染色されたゲルの場合は、直接 10%メタノール/7%酢酸 200ml 中で30分間以上振盪する
。
9-8. 洗浄液を捨ててSYPRO Ruby 染色液(ラージゲルの場合 100 ml、ミニゲルでは 50 ml
以上)を注ぎ、アルミホイルで遮光して、室温で 1.5 時間〜2時間振盪する。
9-9. SYPRO Ruby
染色液も数回再利用できるので、元の容器に戻して室温で保存する。
(低温で保存すると沈殿物ができてゲルに細かな斑点がでることがある)
9-10.
10%メタノール/7%酢酸中で30分間振盪して過剰の蛍光色素を除き、さらにMilli-Q水で10分程度リンスした後に、レーザー励起型-
蛍光スキャナー(Bio-RadのPharos FX や
GEのTyphoonなど)、もしくは発光ダイオード励起型-冷却CCDカメラ撮影装置(AnatechのFluoroPhoreStar3000など)
で蛍光画像を取り込む。
(ゲルの長期保存には、7%酢酸を使用する)
10.画像解析とスポットの切り脱し
バイオラッドのPDQuestなどを用いて、おおよそ以下の手順で画像解析を行う。
10-1.バックグラウンドとノイズの除去。
10-2.スポットの検出とスポット形状の正規化(ガウス分布近似法)。
10-3.スポットのマッチング(ランドマークの入力による)。
10-4.スポットの数値化と正規化(全タンパクスポットの定量値総和に対する相対定量値化)。
10-5.指定範囲を越えて増減が見られたスポットの表示。
この後、データベースを行う時は全てのスポットを、分化や腫瘍化などで変化するタンパク質を同定する場合には、変化が見られたスポットを切り出して、
96穴のプレートもしくは、0.5mlのポリエチレンチューブに移す。
切り出し用の装置としては、バイオラッドの Spot Cutterや、アナテックのFluoroPhoreStar 3000 などが便利である。
11.トリプシンによるゲル内消化
試薬の調製
・100 mM ammonium
bicarbonateの調製:ammonium
bicarbonate 0.8 g を Milli-Q 水 100 ml に溶解する。(冷蔵庫で1〜2ヶ月は保存可能)
・還元処理液の調製:1.5 mg の dithiothreitol (DTT) を 1 ml の100 mM ammonium
bicarbonate に溶解する。(用時調製)
・アルキル化処理液の調製:10 mg の iodoacetamide を 1 ml の100 mM ammonium
bicarbonate に溶解する。(用時調製)
・脱色液Aの調製:特級のメタノールと100 mM ammonium bicarbonate を等量混合する。
・脱色液Bの調製:HPLCグレードのアセトニトリルと100 mM ammonium bicarbonate を等量混合する。
・トリプシン保存液の調製:Sequencing grade modified trypsin (Promega V511A) 1
vial (20 μg)をキットに添付されているTrypsin Resuspension
Buffer(もしくは、50 mM 酢酸)0.1 ml で溶解し、トリプシン保存液とする。
(残ったトリプシン保存液は 24μl ずつ分注して凍結保存する。次のトリプシン消化液は保存不可)
・トリプシン消化液の調製:32 サンプル当たり 24 μl のトリプシン保存液を、トリプシン消化バッファー(30%(v/v)
acetonitrile, 50 mM ammonium
bicaronate)で40倍に希釈する。(用時調製)
(トリプシンの濃度は、サンプル中のタンパク質の量に応じて加減すると良い)
消化の操作手順
11-1.還元処理液 0.1 ml 中に30分間浸す。
11-2.アルキル化試薬液 0.1 ml 中に30分間浸す。
11-3.脱色液A 0.1 ml 中に15分間2回浸す。
11-4.脱色液B 0.1 ml 中に10分間3回浸す。
11-5.100% acetonitrile 0.1 ml 中に5分間浸す。
11-6.液を除いて風乾する。(約10分間)
11-7.トリプシン消化液25μl を加え、30℃で一夜インキュベートする。
*電気泳動の途中で十分に還元アルキル化が行われている場合には、11-1 と 11-2 はスキップしても構わいません。
*AXIMA-CFR MALDI-TOF/MS
によるPMF分析を行なう場合には、消化後のゲルからの抽出操作や脱塩の操作は特に必要ありません。
12.MALDI ターゲットプレート(サンプルプレート)への塗布
12-1.11でインキュベーション後のトリプシン消化液 1 μl を、MALDI ターゲットプレートに載せる。
12-2.マトリックス液1 μl を、MALDI ターゲットプレート上で混合する。
12-3.十分に風乾する。
・マトリックス液の調製法:CHCA (α-cyano-4-hydroxycinnamic acid) 10 mg を、50%
acetonitrile, 40% methanol, 0.1% TFA 混合溶媒 1 ml に溶解する。(CHCA
は予め acetone で洗っておくと、MALDI-TOF 質量分析の際の低分子側のノイズを低減することができる)
マトリックス液は、冷蔵庫中で約1ヶ月程度は保存可能。
13.染色されたゲルからPVDF膜へのタンパク質転写(オプション)
膜状でトリプシン消化を行う時には、SYPRO
Ruby染色後のゲルを純水で良く洗った後(30分間3回くらい)、再可溶化バッファー中で15分間インキュベートし、エレクトロブロット装置にセットし
て、PVDF膜に転写する。再可溶化バッファー、転写バッファー、通電条件は下記の通り。
再可溶化バッファー:
0.3%(w/v) Tris, 0.7%(w/v) glycine, 0.2%(w/v) SDS
転写バッファー:
0.3%(w/v) Tris, 1.5%(w/v) glycine, 0.1%(w/v) SDS
通電条件:
4 V/cm で1夜
この条件では、タンパク質とともにSYPRO
RubyもPVDF膜に移りますので、転写後に染め直さなくても、スポットの位置を確認することができます。
(この「染色されたゲルからPVDF膜へのタンパク質転写」法は、"Proteomic analysis of Epstein-Barr
virus-transformed human
B-lymphoblastoid cell lines before and after immortalization"
Electrophoresis, 21(9), 1814-1822 (2000)
に記載しておりますので、そちらを引用して下さい。)